Генодиагностика синдрома Ретта, поиск мутаций в гене MECP2
Синдром Ретта представляет собой социально значимый генетический синдром, приводящий к регрессу психомоторного развития, аутизму, нарушению развития и нередко к развитию эпилепсии у девочек. Заболевание связано с мутациями гена МЕСР2, который расположен на Х-хромосоме. Исследование предназначено для диагностики типичного варианта синдрома Ретта. Исследуются все точечные мутации в кодирующей области гена MECP2.
Синонимы русские
Синдром Ретта, ген МЕСР2, генетическое обследование.
Синонимы английские
Rett Syndrome,MECP2-related disorders, gene MECP2.
Название гена
Ген MECP2.
Локализация гена на хромосоме
Локус Xq28.
Метод исследования
Полимеразная цепная реакция (ПЦР) – Секвенирование гена MECP2.
Какой биоматериал можно использовать для исследования?
Венозную кровь.
Как правильно подготовиться к исследованию?
- Не курить в течение 30 минут до исследования.
Общая информация об исследовании
Классическая форма синдрома Ретта представляет собой генетическое Х-сцепленное заболевание психомоторного развития. Данный синдром считается самой распространенной моногенной причиной отставания в развитии у девочек. Заболевание ассоциировано с различными видами мутаций в гене MECP2.
Клинически синдром Ретта характеризуется коротким периодом стагнации в развитии на 18-м месяце жизни. Основными проявлениями классической формы данного синдрома являются недостаточность речевых и моторных навыков, нарушение походки, стереотипные движения руками, нарушение интеллектуального развития. Кроме этого, заболевание у пациента может проявляться судорожными припадками, бруксизмом, эпизодическим апноэ, атаксией походки, тремором, спастичностью, характеристиками нозологий аутистического спектра. Помимо классической формы синдрома Ретта, мутации в гене MECP2 могут вызывать атипичные варианты с более серьезной симптоматикой и быстрой прогрессией либо с более мягким течением.
Примерно 99,5% мутаций в гене MECP2 возникают de novo, то есть впервые в данной семье. Редко патологическая мутация может передаваться от носителя-матери, у которой симптомы заболевания либо отсутствуют, либо сглажены. Отсутствие мутаций в гене MECP2 не исключает синдром Ретта в связи с возможностью нахождения генетической аберрации в другом регионе генома.
Для чего используется исследование?
В соответствии с международными клиническими рекомендациями, генетическое обследование на синдром Ретта проводится при наличии у пациента клинической симптоматики, характерной для данного заболевания.
Когда назначается исследование?
- Дифференциальный диагноз отставания развития;
- при подозрении на синдром Ретта;
- при дифференциальной диагностике расстройств аутистического спектра;
- при дифференциальной диагностике эпилепсии;
- при когнитивных и нейропсихических нарушениях.

Что означают результаты?
Генетическое обследование является основным методом подтверждения диагноза и основано на определении точечных мутаций в гене MECP2 с помощью секвенирования.
Референсные значения
Не обнаружено точечных мутаций в гене MECP2.
Положительный результат
Обнаружена патологическая мутация в гене MECP2, характерная для синдрома Ретта.
Что может влиять на результат?
Хотя генетический тест является точным методом лабораторной диагностики, время клинических проявлений заболевания (пенетрантность болезни) зависит от внешней среды, индивидуальных генетических факторов. Для оценки характера наследования у детей и родственников, характера развития заболевания в последующем, назначения лечения рекомендуется получить консультацию специалиста.
Важные замечания
- Для получения заключения по результату обследования необходимо проконсультироваться у клинического генетика.

Кто назначает исследование?
Невролог, психиатр, врач-генетик.
Также рекомендуется
[06-437] Анализ крови на аминокислоты (48 показателей)
[16-001] Исследование кариотипа (количественные и структурные аномалии хромосом) по лимфоцитам периферической крови (1 человек)
Литература
- Matijevic T, Knezevic J, Slavica M, Pavelic J, Rett Syndrome: From the Gene to the Disease. Eur Neurol 2009;61:3-10.
- Rett syndrome: Revised diagnostic criteria and nomenclature. Ann Neurol 2010;68:944 – 50.
- Christodoulou J, Ho G. MECP2-Related Disorders. 2001 Oct 3 [Updated 2012 Jun 28]. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2018.
Синдром Ретта: клинический случай | Бурлуцкая
1. Percy A.K. Progress in Rett Syndrome: from discovery to clinical trials. Wien. Med. Wochenschr. 2016; 166(11-12): 325-332. DOI: 10.1007/s10354-016-0491-9
2016; 166(11-12): 325-332. DOI: 10.1007/s10354-016-0491-9
2. Ворсанова С.Г., Юров Ю.Б., Воинова В.Ю., Юров И.Ю. Синдром Ретта в России и зарубежом: научный исторический обзор. Российский вестник перинатологии и педиатрии. 2020; 65:(3): 25-31. DOI: 10.21508/1027-4065-2020-653-25-31
3. Воинова В.Ю., Ворсанова С.Г., Юров Ю.Б., Юров И.Ю. Алгоритм диагностики Х-сцепленных форм умственной отсталости у детей. Российский вестник перинатологии и педиатрии. 2016; 61(5): 34-41. DOI: 10.21508/1027-4065-201661-5-34-41
4. Gold W.A., Krishnarajy R., Ellaway C., Christodoulou J. Rett Syndrome: A Genetic Update and Clinical Review Focusing on Comorbidities. ACS Chem. Neuro-sci. 2018; 9(2): 167-176. DOI: 10.1021/acschemneuro.7b00346
5. Leonard H., Cobb S., Downs J. Clinical and biological progress over 50 years in Rett syndrome. Nat. Rev. Neurol. 2017; 13(1): 37-51. DOI: 10.1038/nrneu-rol.2016.186
6. Fehr S., Downs J., Ho G., de Klerk N., Forbes D., Christodoulou J., Williams S. , Leonard H. Functional abilities in children and adults with the CDKL5 disorder. Am. J. Med. Genet. A. 2016; 170(11): 2860-2869. DOI: 10.1002/ajmg.a.37851
, Leonard H. Functional abilities in children and adults with the CDKL5 disorder. Am. J. Med. Genet. A. 2016; 170(11): 2860-2869. DOI: 10.1002/ajmg.a.37851
7. Lyst M.J., Bird A. Rett syndrome: a complex disorder with simple roots. Nat. Rev. Genet. 2015; 16(5): 261275. DOI: 10.1038/nrg3897
8. Qiu Z. Deciphering MECP2-associated disorders: disrupted circuits and the hope for repair. Curr. Opin. Neurobiol. 2018; 48: 30-36. DOI: 10.1016/j.conb.2017.09.004
9. Lopes F., Barbosa M., Ameur A., Soares G., de Sa J., Dias A.I., Oliveira G., Cabral P., Temudo T., Calado E., Cruz I.F., Vieira J.P., Oliveira R., Esteves S., Sauer S., Jonasson I., Syvanen A.C., Gyllensten U., Pinto D., Maciel P. Identification of novel genetic causes of Rett syndrome-like phenotypes. J. Med. Genet. 2016; 53(3): 190-199. DOI: 10.1136/jmedgenet-2015-103568
10. Малинина Е.В., Забозлаева И.В. Синдром Ретта: трудности диагностики (клинико-психопатологические аспекты). Русский журнал детской неврологии. 2016; 11(3): 49-56. DOI: 10.17650/20738803-2016-11-3-49-56
2016; 11(3): 49-56. DOI: 10.17650/20738803-2016-11-3-49-56
11. Fonzo M., Sirico F., Corrado B. Evidence-Based Physical Therapy for Individuals with Rett Syndrome: A Systematic Review. Brain. Sci. 2020; 10(7): 410. DOI: 10.3390/brainsci10070410
12. Gogliotti R.G., Niswender C.M. A Coordinated Attack: Rett Syndrome Therapeutic Development. Trends. Pharmacol. Sci. 2019; 40(4): 233-236. DOI: 10.1016/j.tips.2019.02.007
13. Sajan S.A., Jhangiani S.N., Muzny D.M., Gibbs R.A., Lupski J.R., Glaze D.G., Kaufmann W.E., Skinner S.A., Annese F., Friez M.J., Lane J., Percy A.K., Neul J.L. Enrichment of mutations in chromatin regulators in people with Rett syndrome lacking mutations in MECP2. Genet. Med. 2017; 19(1): 13-19. DOI: 10.1038/gim.2016.42
14. Townend G.S., Ehrhart F., van Kranen H.J., Wilkinson M., Jacobsen A., Roos M., Willighagen E.L., van Enckevort D., Evelo C.T., Curfs L.M.G.. MECP2 variation in Rett syndrome-An overview of current coverage of genetic and phenotype data within existing databases. Hum. Mutat. 2018; 39(7): 914924. DOI: 10.1002/humu.23542
Hum. Mutat. 2018; 39(7): 914924. DOI: 10.1002/humu.23542
15. Ehrhart F., Sangani N.B., Curfs L.M.G. Current developments in the genetics of Rett and Rett-like syndrome. Curr. Opin. Psychiatry. 2018; 31(2): 103-108. DOI: 10.1097/YCO.0000000000000389
Синдром Ретта | Национальный институт неврологических расстройств и инсульта
Что такое синдром Ретта?
Синдром Ретта — это нарушение развития нервной системы. Характеризуется типичным ранним ростом и развитием, за которым затем следует:
- Замедление развития
- Потеря подвижности или функции рук
- Характерные движения рук
- Замедленный рост мозга и головы
- Проблемы с ходьбой, ходьба на носочках или походка с широкой опорой
- Изъятия
- Когнитивные проблемы
- Проблемы пищеварения
- Проблемы с двигательными функциями, включая речь и контроль движений глаз (апраксия)
- Затрудненное дыхание во время бодрствования, включая задержку дыхания, гипервентиляцию и глотание воздуха
Другие симптомы могут включать проблемы со сном, скрежетание зубами и затрудненное жевание.
Возраст начала заболевания, тяжесть симптомов и характер прогрессирования синдрома Ретта варьируются от ребенка к ребенку. В то время как дети, у которых развивается синдром Ретта, изначально растут и развиваются, как и ожидалось, даже в раннем младенчестве и дошкольном возрасте часто наблюдаются тонкие различия, такие как:
- Потеря мышечного тонуса (гипотония)
- Проблемы с кормлением
- Подергивание при движениях конечностей
- Проблемы с ползанием или ходьбой
- Меньше зрительного контакта
Потеря функционального использования рук сопровождается компульсивными движениями рук, такими как заламывание рук. Появление этих симптомов иногда бывает внезапным.
У кого больше шансов заболеть синдромом Ретта?
Синдром Ретта может быть у любой расовой или этнической группы. Синдром Ретта чаще всего поражает девочек, хотя мальчики также (редко) страдают. Мальчики обычно болеют тяжелее, чем девочки.
Почти все случаи синдрома Ретта вызваны мутацией метилового CpG-связывающего белка 2, или гена MECP2 (произносится как meck-pea-two). Ген MECP2 содержит инструкции по синтезу белка, называемого метилцитозин-связывающим белком 2 (MeCP2), который необходим для развития мозга и действует как один из многих биохимических переключателей, активирующих и деактивирующих функции генов. Поскольку ген MECP2 не функционирует должным образом у людей с синдромом Ретта, у них может быть слишком мало MeCP2, или MeCP2, который у них есть, не работает должным образом.
Ген MECP2 содержит инструкции по синтезу белка, называемого метилцитозин-связывающим белком 2 (MeCP2), который необходим для развития мозга и действует как один из многих биохимических переключателей, активирующих и деактивирующих функции генов. Поскольку ген MECP2 не функционирует должным образом у людей с синдромом Ретта, у них может быть слишком мало MeCP2, или MeCP2, который у них есть, не работает должным образом.
Не у всех, у кого есть мутация MECP2, есть синдром Ретта. Ученые считают, что некоторые случаи могут быть вызваны частичными делециями генов, мутациями в других частях гена MECP2 или дополнительными генами, которые еще не идентифицированы. Генетические факторы и факторы окружающей среды могут способствовать различиям в тяжести и типах симптомов, обнаруживаемых у людей с синдромом Ретта. К ним могут относиться: где в гене расположена мутация MECP2, как взаимодействуют половые хромосомы людей, и другие гены могут ухудшить симптомы или защитить от последствий мутации.
Хотя синдром Ретта является генетическим заболеванием, менее 1% зарегистрированных случаев передается от одного поколения к другому. Большинство случаев являются спонтанными, что означает, что мутация происходит случайно. Однако в некоторых семьях людей, страдающих синдромом Ретта, другие члены семьи имеют мутацию в гене MECP2. Семьи, в которых уже выявлена мутация MECP2, могут пройти генетическое тестирование, чтобы определить, являются ли они носителями этого заболевания.
Как диагностируется и лечится синдром Ретта?
Диагностика синдрома Ретта
Врачи диагностируют синдром Ретта, наблюдая признаки и симптомы в период раннего роста и развития ребенка и проводя постоянную оценку физического и неврологического статуса ребенка. Ученые разработали генетический тест для обнаружения мутаций MECP2, чтобы дополнить наблюдения и диагностику врачей. Чтобы подтвердить диагноз синдрома Ретта, семьям следует проконсультироваться с детским неврологом, клиническим генетиком или педиатром по развитию.
Лечение синдрома Ретта
Хотя лекарства от синдрома Ретта не существует, в 2023 году Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) одобрило новый препарат Трофинетид для лечения синдрома Ретта у детей в возрасте двух лет и старше. Трофинетид работает, уменьшая отек головного мозга, увеличивая количество защитного белка в мозге и останавливая чрезмерную активность некоторых клеток.
Другие методы лечения сосредоточены на лечении конкретных симптомов или поведения, наблюдаемых при синдроме Ретта. Лекарства могут помочь при проблемах с дыханием и движениями, а также при судорогах.
Людей с синдромом Ретта следует регулярно обследовать на наличие сколиоза (искривления позвоночника) и проблем с сердцем. Трудотерапия может помочь детям развить навыки, необходимые для повседневной деятельности, такой как одевание, прием пищи и рисование, в то время как физиотерапия и водная терапия (гидротерапия) могут помочь с подвижностью.
Некоторым детям может потребоваться специальное оборудование и вспомогательные средства, такие как корсеты для позвоночника, шины для рук и запястий, а также пищевая поддержка для поддержания адекватного веса. Детям с синдромом Ретта может потребоваться дополнительная помощь в школе и социальная поддержка.
Многие люди с синдромом Ретта доживают до среднего возраста и старше, несмотря на симптомы. Поскольку заболевание встречается редко, очень мало известно о долгосрочном прогнозе и ожидаемой продолжительности жизни.
Какие последние новости о синдроме Ретта?
Национальные институты здравоохранения (NIH), включая NINDS и другие, поддерживают исследования синдрома Ретта. Понимание причины необходимо для разработки новых методов лечения конкретных симптомов, а также для улучшения диагностических инструментов. Открытие MECP2 в 1999 обеспечивает основу для дальнейших генетических исследований и позволяет использовать недавно разработанные модели животных, такие как трансгенные мыши с дефицитом MECP2. Эти модели имеют неврологические аномалии, которые могут быть устранены путем активации гена MECP2 в более позднем возрасте.
Эти модели имеют неврологические аномалии, которые могут быть устранены путем активации гена MECP2 в более позднем возрасте.
Ученые изучают мутации в гене MECP2 у людей с синдромом Ретта, чтобы узнать о функции и дисфункции белка MeCP2. Информация из этого исследования расширит наше понимание расстройства и может привести к новым методам лечения. Другие исследования направлены на выявление затронутых молекулярных путей, разработку моделей расстройства на животных и разработку терапии на ранних стадиях.
Некоторые исследователи предполагают, что конкретный тип мутации в гене MECP2 влияет на тяжесть симптомов синдрома Ретта. Ведутся исследования, чтобы понять, какие мутации и гены способствуют разнообразию симптомов и тяжести этого расстройства. Одно исследование естественной истории, финансируемое NIH, также должно предоставить новую информацию по этим темам.
Ученые знают, что отсутствие правильно функционирующего белка MeCP2 нарушает функцию зрелых клеток мозга, но они не знают точных механизмов, с помощью которых это происходит. Исследователи пытаются найти другие генетические переключатели, которые действуют аналогично белку MeCP2. Как только они узнают, как работает белок, и найдут похожие переключатели, они смогут разработать методы лечения, которые смогут заменить неисправный переключатель. Другой результат может включать манипулирование другими биохимическими путями для компенсации неисправного гена MECP2, тем самым предотвращая прогрессирование расстройства. В настоящее время проводятся ранние клинические испытания генной терапии синдрома Ретта.
Исследователи пытаются найти другие генетические переключатели, которые действуют аналогично белку MeCP2. Как только они узнают, как работает белок, и найдут похожие переключатели, они смогут разработать методы лечения, которые смогут заменить неисправный переключатель. Другой результат может включать манипулирование другими биохимическими путями для компенсации неисправного гена MECP2, тем самым предотвращая прогрессирование расстройства. В настоящее время проводятся ранние клинические испытания генной терапии синдрома Ретта.
Исследователи также пытаются найти другие гены, которые могут быть связаны с синдромом Ретта. Некоторые исследования помогли сузить поиск этих генов, но многое еще неизвестно о том, как эти гены могут вызывать расстройство или способствовать ему.
file-medical
Узнайте о клинических испытаниях
Клинические испытания — это исследования, которые позволяют нам больше узнать о заболеваниях и улучшить лечение. Они могут помочь пациентам узнать о новых и предстоящих вариантах лечения.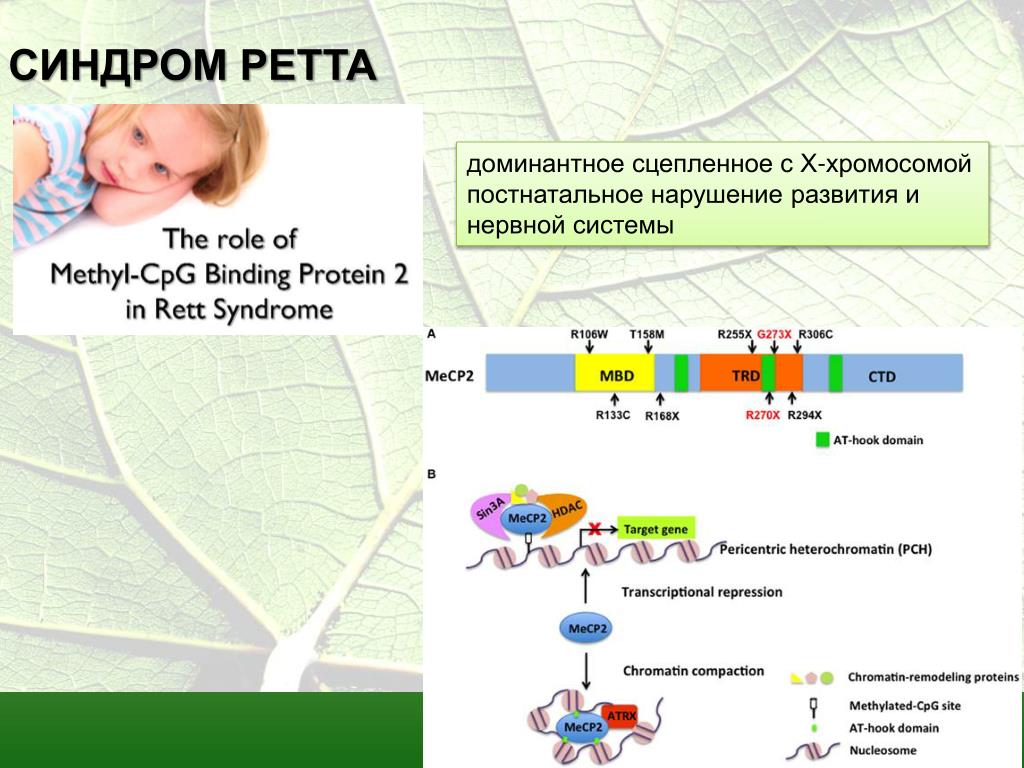
Как я или мой близкий человек могу помочь улучшить уход за людьми с синдромом Ретта?
Рассмотрите возможность участия в клинических испытаниях, чтобы клиницисты и ученые могли больше узнать о синдроме Ретта и связанных с ним расстройствах. В клинических исследованиях участвуют люди-добровольцы, чтобы помочь исследователям узнать больше о расстройстве и, возможно, найти более эффективные способы безопасного выявления, лечения или предотвращения болезни.
Нужны все типы добровольцев — здоровые или, возможно, больные или больные — всех возрастов, полов, рас и этнических групп, чтобы гарантировать, что результаты исследования применимы к как можно большему количеству людей, и что лечение будет безопасны и эффективны для всех, кто будет их использовать.
Для получения информации об участии в клинических исследованиях посетите сайт NIH Clinical Research Trials and You. Узнайте о клинических испытаниях, в которых в настоящее время ищут людей с синдромом Ретта, на сайте Clinicaltrials.
Где я могу найти дополнительную информацию о синдроме Ретта?
Информация может быть получена от следующих организаций:
Информационный центр генетических и редких заболеваний (GARD)
Тел.: 888-205-2311Международный фонд синдрома Ретта
Телефон: 513-874-1298 или 800-818-7388Национальный институт детского здоровья и человеческого развития (NICHD)
Телефон: 301-496-5133Национальный институт психического здоровья (NIMH)
Телефон: 301- 443-4513 или 866-415-8051 или 301-443-8431Национальная организация по редким заболеваниям (NORD)
Телефон: 203-744-0100 или 800-999-6673 или 844-259-7178 Испанский
Синдром Ретта: MedlinePlus Genetics
Описание
Синдром Ретта — это заболевание головного мозга, которое встречается почти исключительно у девочек. Наиболее распространенная форма заболевания известна как классический синдром Ретта. После рождения девочки с классическим синдромом Ретта в течение 6–18 месяцев кажутся нормально развитыми, прежде чем у них разовьются серьезные проблемы с речью и общением, обучением, координацией и другими функциями мозга. В раннем детстве больные девочки перестают целенаправленно пользоваться руками и начинают многократно заламывать руки, умываться или хлопать в ладоши. Они, как правило, растут медленнее, чем другие дети, и около трех четвертей имеют небольшой размер головы (микроцефалия). Другие признаки и симптомы, которые могут развиться, включают аномалии дыхания, срыгивание или слюнотечение, необычные движения глаз, такие как напряженный взгляд или чрезмерное моргание, холодные руки и ноги, раздражительность, нарушения сна, судороги и аномальное искривление позвоночника из стороны в сторону. (сколиоз).
В раннем детстве больные девочки перестают целенаправленно пользоваться руками и начинают многократно заламывать руки, умываться или хлопать в ладоши. Они, как правило, растут медленнее, чем другие дети, и около трех четвертей имеют небольшой размер головы (микроцефалия). Другие признаки и симптомы, которые могут развиться, включают аномалии дыхания, срыгивание или слюнотечение, необычные движения глаз, такие как напряженный взгляд или чрезмерное моргание, холодные руки и ноги, раздражительность, нарушения сна, судороги и аномальное искривление позвоночника из стороны в сторону. (сколиоз).
Исследователи описали несколько вариантов или атипичных форм синдрома Ретта, которые могут быть более легкими или более тяжелыми, чем классическая форма.
Синдром Ретта является частью спектра заболеваний с одной и той же генетической причиной. Другие расстройства в этом спектре включают синдром PPM-X, синдром дупликации MECP2 и тяжелую неонатальную энцефалопатию, связанную с MECP2 . Эти другие условия могут повлиять на мужчин.
Эти другие условия могут повлиять на мужчин.
Частота
Это состояние затрагивает примерно 1 из 9от 000 до 10 000 женщин.
Причины
Мутации в гене MECP2 лежат в основе почти всех случаев классического синдрома Ретта и некоторых вариантов его состояния. Этот ген дает инструкции для создания белка (MeCP2), который имеет решающее значение для нормальной работы мозга. Хотя точная функция белка MeCP2 неясна, вероятно, он участвует в поддержании связей (синапсов) между нервными клетками (нейронами). Он также может быть необходим для нормального функционирования других типов клеток головного мозга.
Считается, что белок MeCP2 помогает регулировать активность генов в головном мозге. Этот белок может также контролировать производство различных версий определенных белков в клетках головного мозга. Мутации в гене MECP2 изменяют белок MeCP2 или приводят к выработке меньшего количества белка, что, по-видимому, нарушает нормальную функцию нейронов и других клеток головного мозга. В частности, исследования показывают, что изменения в белке MeCP2 могут снижать активность определенных нейронов и нарушать их способность общаться друг с другом. Неясно, как эти изменения приводят к специфическим чертам синдрома Ретта.
В частности, исследования показывают, что изменения в белке MeCP2 могут снижать активность определенных нейронов и нарушать их способность общаться друг с другом. Неясно, как эти изменения приводят к специфическим чертам синдрома Ретта.
Было обнаружено, что несколько состояний с признаками и симптомами, перекрывающими симптомы синдрома Ретта, являются результатом мутаций в других генах. Эти состояния, включая синдром FOXG1 и дефицит CDKL5, ранее считались вариантными формами синдрома Ретта. Однако врачи и исследователи выявили некоторые важные различия между состояниями, поэтому теперь их обычно считают отдельными расстройствами.
Наследство
Более 9У 9 процентов людей с синдромом Ретта в их семье нет анамнеза этого расстройства. Многие из этих случаев являются результатом новых мутаций в гене MECP2 .
Описано несколько семей, в которых более одного больного члена семьи. Эти случаи помогли исследователям определить, что классический синдром Ретта и варианты, вызванные мутациями гена MECP2 , имеют Х-сцепленный доминантный тип наследования. Состояние считается сцепленным с Х-хромосомой, если мутировавший ген, вызывающий заболевание, расположен на Х-хромосоме, одной из двух половых хромосом. Наследование является доминантным, если одной копии измененного гена в каждой клетке достаточно, чтобы вызвать заболевание.
Состояние считается сцепленным с Х-хромосомой, если мутировавший ген, вызывающий заболевание, расположен на Х-хромосоме, одной из двух половых хромосом. Наследование является доминантным, если одной копии измененного гена в каждой клетке достаточно, чтобы вызвать заболевание.
Самцы с мутациями в гене MECP2 часто умирают в младенчестве. Однако у небольшого числа мужчин с генетическим изменением, связанным с MECP2 , развились признаки и симптомы, сходные с симптомами синдрома Ретта, включая умственную отсталость, судороги и проблемы с движением. У мужчин это состояние описывается как тяжелая неонатальная энцефалопатия, связанная с MECP2 . Признаки и симптомы у некоторых мужчин с мутацией гена MECP2 находятся в более легкой форме.
Другие названия этого состояния
- Аутизм-деменция-атаксия-синдром потери целенаправленного использования рук
- Синдром Ретта
- Болезнь Ретта
- Синдром Ретта
- РТТ
Дополнительная информация и ресурсы
Информация о генетическом тестировании
- Реестр генетического тестирования: синдром Ретта
Информационный центр генетических и редких заболеваний
- Атипичный синдром Ретта
- Синдром Ретта
Ресурсы поддержки пациентов и защиты интересов
- Информационный поиск по болезням
- Национальная организация редких заболеваний (NORD)
Научные исследования от ClinicalTrials.
 gov
gov- ClinicalTrials.gov
Каталог генов и болезней от OMIM
- СИНДРОМ РЕТТА
Научные статьи в PubMed
- ПабМед
Ссылки
- Чарур М., Зогби Х.И. История синдрома Ретта: от клиники до нейробиология. Нейрон. 2007 8 ноября; 56 (3): 422-37. doi: 10.1016/j.neuron.2007.10.001. Цитата в PubMed
- Эрхарт Ф., Коорт С.Л., Чирилло Э., Смитс Э., Эвело КТ, Керфс Л.М. Синдром Ретта — биологические пути, ведущие от MECP2 к фенотипам расстройств. Сирота J Редкий Дис. 2016 25 ноября; 11 (1): 158. doi: 10.1186/s13023-016-0545-5. Цитирование в PubMed или бесплатная статья в PubMed Central
- Gold WA, Krishnarajy R, Ellaway C, Christodoulou J. Синдром Ретта: генетический
Обновление и клинический обзор с акцентом на сопутствующие заболевания. ACS Chem Neurosci. 2018 февраль
21;9(2):167-176. doi: 10.1021/acschemneuro.7b00346. Epub 2017, 15 декабря.

