Синдром Ретта: клинический случай | Бурлуцкая
1. Percy A.K. Progress in Rett Syndrome: from discovery to clinical trials. Wien. Med. Wochenschr. 2016; 166(11-12): 325-332. DOI: 10.1007/s10354-016-0491-9
2. Ворсанова С.Г., Юров Ю.Б., Воинова В.Ю., Юров И.Ю. Синдром Ретта в России и зарубежом: научный исторический обзор. Российский вестник перинатологии и педиатрии. 2020; 65:(3): 25-31. DOI: 10.21508/1027-4065-2020-653-25-31
3. Воинова В.Ю., Ворсанова С.Г., Юров Ю.Б., Юров И.Ю. Алгоритм диагностики Х-сцепленных форм умственной отсталости у детей. Российский вестник перинатологии и педиатрии. 2016; 61(5): 34-41. DOI: 10.21508/1027-4065-201661-5-34-41
4. Gold W.A., Krishnarajy R., Ellaway C., Christodoulou J. Rett Syndrome: A Genetic Update and Clinical Review Focusing on Comorbidities. ACS Chem. Neuro-sci. 2018; 9(2): 167-176. DOI: 10.1021/acschemneuro.7b00346
5. Leonard H., Cobb S., Downs J. Clinical and biological progress over 50 years in Rett syndrome. Nat. Rev. Neurol. 2017; 13(1): 37-51. DOI: 10.1038/nrneu-rol.2016.186
Nat. Rev. Neurol. 2017; 13(1): 37-51. DOI: 10.1038/nrneu-rol.2016.186
6. Fehr S., Downs J., Ho G., de Klerk N., Forbes D., Christodoulou J., Williams S., Leonard H. Functional abilities in children and adults with the CDKL5 disorder. Am. J. Med. Genet. A. 2016; 170(11): 2860-2869. DOI: 10.1002/ajmg.a.37851
7. Lyst M.J., Bird A. Rett syndrome: a complex disorder with simple roots. Nat. Rev. Genet. 2015; 16(5): 261275. DOI: 10.1038/nrg3897
8. Qiu Z. Deciphering MECP2-associated disorders: disrupted circuits and the hope for repair. Curr. Opin. Neurobiol. 2018; 48: 30-36. DOI: 10.1016/j.conb.2017.09.004
9. Lopes F., Barbosa M., Ameur A., Soares G., de Sa J., Dias A.I., Oliveira G., Cabral P., Temudo T., Calado E., Cruz I.F., Vieira J.P., Oliveira R., Esteves S., Sauer S., Jonasson I., Syvanen A.C., Gyllensten U., Pinto D., Maciel P. Identification of novel genetic causes of Rett syndrome-like phenotypes. J. Med. Genet. 2016; 53(3): 190-199. DOI: 10.1136/jmedgenet-2015-103568
10. Малинина Е.В., Забозлаева И.В. Синдром Ретта: трудности диагностики (клинико-психопатологические аспекты). Русский журнал детской неврологии. 2016; 11(3): 49-56. DOI: 10.17650/20738803-2016-11-3-49-56
Малинина Е.В., Забозлаева И.В. Синдром Ретта: трудности диагностики (клинико-психопатологические аспекты). Русский журнал детской неврологии. 2016; 11(3): 49-56. DOI: 10.17650/20738803-2016-11-3-49-56
11. Fonzo M., Sirico F., Corrado B. Evidence-Based Physical Therapy for Individuals with Rett Syndrome: A Systematic Review. Brain. Sci. 2020; 10(7): 410. DOI: 10.3390/brainsci10070410
12. Gogliotti R.G., Niswender C.M. A Coordinated Attack: Rett Syndrome Therapeutic Development. Trends. Pharmacol. Sci. 2019; 40(4): 233-236. DOI: 10.1016/j.tips.2019.02.007
13. Sajan S.A., Jhangiani S.N., Muzny D.M., Gibbs R.A., Lupski J.R., Glaze D.G., Kaufmann W.E., Skinner S.A., Annese F., Friez M.J., Lane J., Percy A.K., Neul J.L. Enrichment of mutations in chromatin regulators in people with Rett syndrome lacking mutations in MECP2. Genet. Med. 2017; 19(1): 13-19. DOI: 10.1038/gim.2016.42
14. Townend G.S., Ehrhart F., van Kranen H.J., Wilkinson M., Jacobsen A., Roos M., Willighagen E. L., van Enckevort D., Evelo C.T., Curfs L.M.G.. MECP2 variation in Rett syndrome-An overview of current coverage of genetic and phenotype data within existing databases. Hum. Mutat. 2018; 39(7): 914924. DOI: 10.1002/humu.23542
L., van Enckevort D., Evelo C.T., Curfs L.M.G.. MECP2 variation in Rett syndrome-An overview of current coverage of genetic and phenotype data within existing databases. Hum. Mutat. 2018; 39(7): 914924. DOI: 10.1002/humu.23542
15. Ehrhart F., Sangani N.B., Curfs L.M.G. Current developments in the genetics of Rett and Rett-like syndrome. Curr. Opin. Psychiatry. 2018; 31(2): 103-108. DOI: 10.1097/YCO.0000000000000389
Два препарата снимают признаки синдрома Ретта в органоидах мозга
Подготовила
Екатерина Петрова
Исследователи из Калифорнийского университета в Сан-Диего с коллабораторами использовали органоиды человеческого мозга с мутациями в гене МЕСР2 для поиска лечения синдрома Ретта. Они нашли два соединения, восстанавливающие синаптическую активность нейронов с дефицитом МЕСР2.
Подготовила
Екатерина Петрова
Синдром Ретта — редкое генетическое заболевание, поражающее преимущественно женщин — связан с мутацией сцепленного с Х-хромосомой гена МЕСР2, который кодирует белок эпигенетической регуляции MeCP2.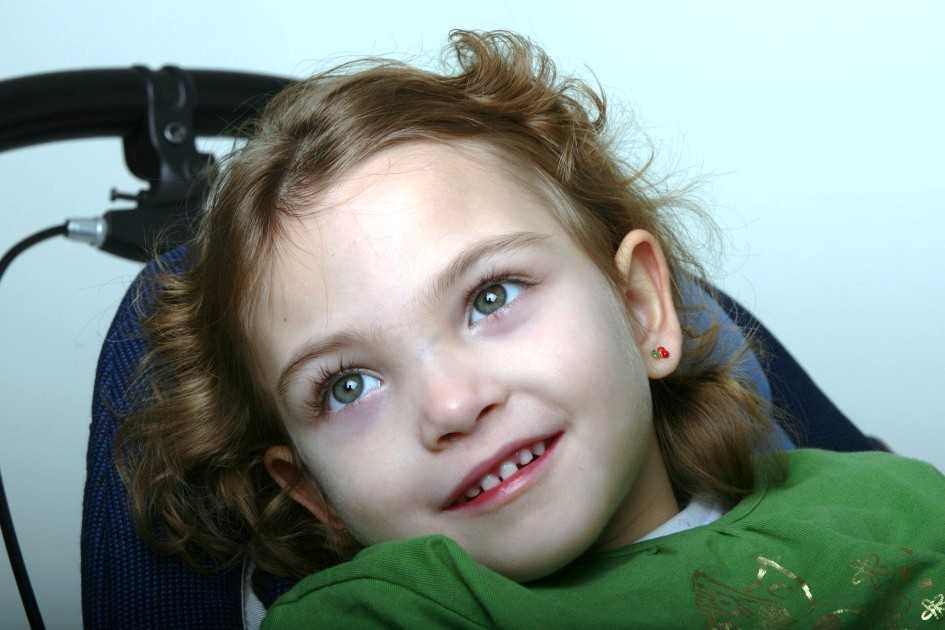 Дефицит этого белка приводит к нарушениям развития нервной системы и регрессу приобретенных в младенчестве навыков. Нейробиологические механизмы связи мутаций
Дефицит этого белка приводит к нарушениям развития нервной системы и регрессу приобретенных в младенчестве навыков. Нейробиологические механизмы связи мутаций
Разработка терапии синдрома Ретта и других нарушений развития нервной системы до сих пор были ограничены отсутствием наиболее подходящих моделей. В новой работе в качестве платформы для скрининга и выявления препаратов, способных специфически устранять нейроцитопатологию, вызванную нокаутом МЕСР2, ученые использовали индуцированные плюрипотентные стволовые клетки (ИПСК) от пациентов с синдромом Ретта. Исследователи не только дифференцировали ИПСК в нейроны, но и вырастили нейросферы и кортикальные органоиды. Чтобы сымитировать мозаичное строение тканей, характерное для женского организма, команда сконструировала органоиды мозга из смеси нейронов с дефицитом MECP2 и нормальных клеток.
«Мутация гена, вызывающая синдром Ретта, была обнаружена несколько десятилетий назад, но прогресс в ее лечении отставал отчасти из-за того, что исследования на мышах не были перенесены на людей. Это исследование было вызвано потребностью в модели, которая лучше имитирует человеческий мозг», — говорит главный автор работы Алиссон Муотри, профессор педиатрии и клеточной и молекулярной медицины в Медицинской школе Калифорнийского университета в Сан-Диего.
Отсутствие MECP2 послужило причиной морфологических и физиологических изменений в органоидах. Так, изменилась форма нейронов, паттерны экспрессии генов, производство нейротрансмиттеров, формирование синапсов, снизилась активность кальция и, как следствие, активность нейронной сети.
В поисках средства для компенсации отсутствия МЕСР2
авторы проверили на органоидах мозга 14 лекарств, противодействующих патологиям синапсов и нейротрансмиттеров. Из всех кандидатов наиболее успешными оказались два: нефирацетам и РНА 543613.
Авторы считают, что полученные результаты дают предпосылки к клиническим испытаниям нефирацетама и РНА 543613 на пациентах с нарушениями психического развития, обусловленных дефицитом МЕСР2.
Источник
Trujillo C. A., et al. // Pharmacological reversal of synaptic and network pathology in human MECP2‐KO neurons and cortical organoids. // EMBO Molecular Medicine (2020), published 08 December 2020; DOI:
// EMBO Molecular Medicine (2020), published 08 December 2020; DOI:
10.15252/emmm.202012523
Цитата по
пресс-релизу
Технологии Неврология Клеточная биология
Синдром Ретта | Национальный институт неврологических расстройств и инсульта
ПОДЕЛИТЬСЯ:
Какие исследования проводятся?
Исследования, финансируемые Национальным институтом неврологических расстройств и инсульта (NINDS) и другими институтами Национальных институтов здравоохранения, включают проекты, направленные на лучшее понимание причины синдрома Ретта, разработку новых методов лечения конкретных симптомов и предоставление более эффективных методов диагностики . Исследователи NINDS изучают основные механизмы в мозге, которые способствуют развитию и прогрессированию синдрома Ретта, и то, как их можно обратить вспять. Это исследование также поможет разработать новые методы лечения синдрома Ретта и других расстройств, которые имеют схожие клеточные механизмы, включая аутизм.
Определение
Определение
ЛечениеЛечение
ПрогнозПрогноз
Клинические испытанияКлинические испытания
ОрганизацииОрганизации
ПубликацииПубликации
ОпределениеОпределение
Синдром Ретта — это редкое детское неврологическое заболевание и нарушение развития, которым страдают почти исключительно женщины.
• потеря мышечного тонуса
• замедление развития
• трудности с кормлением
• судорожные движения рук и ног
• снижение зрительного контакта и взгляда.
Затем ребенок начинает терять или регрессировать ранее приобретенные навыки, в том числе:
• способность общаться и говорить
• целенаправленное использование рук
• способность ходить.
Другие симптомы могут включать:
• замедление роста
• судороги
• умственную отсталость
• проблемы с дыханием
• сколиоз (искривление позвоночника)
Симптомы обычно стабилизируются в возрасте 3–5 лет. Социальные взаимодействия продолжают улучшаться во взрослом возрасте, но двигательная функция и движение постепенно ухудшаются, а мышцы становятся все более слабыми.
Читать далее Читать меньше
ЛечениеЛечение
Синдром Ретта неизлечим. Лечение фокусируется на симптомах расстройства наряду с поддерживающей терапией. Лекарства могут облегчить неровности дыхания, затруднения движений и судороги. Трудотерапия может помочь детям развить навыки, необходимые для выполнения таких действий, как одевание и кормление. Физиотерапия и другие формы терапии могут увеличить подвижность. Некоторым детям может потребоваться специальное оборудование, такое как скобы для лечения сколиоза, шины для изменения движений рук и программы питания. В некоторых случаях могут потребоваться специальные академические, социальные, профессиональные и вспомогательные услуги.
Лечение фокусируется на симптомах расстройства наряду с поддерживающей терапией. Лекарства могут облегчить неровности дыхания, затруднения движений и судороги. Трудотерапия может помочь детям развить навыки, необходимые для выполнения таких действий, как одевание и кормление. Физиотерапия и другие формы терапии могут увеличить подвижность. Некоторым детям может потребоваться специальное оборудование, такое как скобы для лечения сколиоза, шины для изменения движений рук и программы питания. В некоторых случаях могут потребоваться специальные академические, социальные, профессиональные и вспомогательные услуги.
Читать далее Читать меньше
ПрогнозПрогноз
Течение синдрома Ретта, включая возраст начала заболевания и тяжесть симптомов, варьируется от ребенка к ребенку. Несмотря на трудности с симптомами, многие люди с синдромом Ретта продолжают жить в среднем возрасте и дольше. Поскольку расстройство встречается редко, требуется больше информации для оценки долгосрочного прогноза и ожидаемой продолжительности жизни.
Читать далее Читать меньше
Клинические испытанияКлинические испытания
Клинические испытания NINDS в США и во всем мире
Синдром Ретта — NHS
По оценкам, им страдает примерно 1 из 12 000 девочек, рождающихся каждый год, и лишь изредка он встречается у мальчиков.
Признаки и симптомы
Некоторые дети с синдромом Ретта страдают более тяжело, чем другие. Кроме того, возраст, в котором впервые появляются симптомы, варьируется от ребенка к ребенку.
У ребенка могут отсутствовать все симптомы синдрома Ретта, и их симптомы могут меняться по мере взросления.
Синдром Ретта описан в 4 стадиях, хотя симптомы часто перекрываются между каждой стадией. Ниже приведены основные характеристики каждой стадии:
Стадия 1: ранние признаки
Сначала кажется, что ребенок развивается и растет нормально в течение как минимум 6 месяцев. Могут быть тонкие признаки синдрома Ретта до того, как у ребенка будет распознано наличие проблемы (особенно задним числом).
Стадия 1 иногда описывается как «застой». Симптомы включают:
- низкий мышечный тонус (гипотония)
- трудности с кормлением
- необычные, повторяющиеся движения рук или судорожные движения конечностей
- задержка в развитии речи интерес к игрушкам
Эти симптомы обычно начинаются в возрасте от 6 до 18 месяцев и часто длятся несколько месяцев, хотя могут сохраняться в течение года и более.
Стадия 1 часто может оставаться незамеченной, поскольку изменения происходят постепенно и могут быть незаметными.
Стадия 2: регрессия
На стадии 2, известной как «регрессия» или «стадия быстрого разрушения», ребенок начинает терять некоторые из своих способностей. Эта стадия обычно начинается в возрасте от 1 до 4 лет и может длиться в любое время от 2 месяцев до более чем 2 лет.
У ребенка постепенно или внезапно начнут развиваться серьезные проблемы с общением и речью, памятью, подвижностью, координацией и другими функциями мозга. Некоторые характеристики и поведение сходны с таковыми при аутизме.
Признаки на этом этапе включают:
- потерю способности целенаправленно использовать руки – часто трудно контролировать повторяющиеся движения рук, которые включают скручивание, умывание, хлопание в ладоши или постукивание
- периоды беспокойства, раздражительности и иногда крика «нет очевидная причина
- социальная изоляция – потеря интереса к людям и избегание зрительного контакта
- неустойчивость и неловкость при ходьбе
- проблемы со сном
- замедление роста головы
- трудности с приемом пищи, жеванием или глотанием, а иногда и запоры, которые могут вызывать боли в животе
Позже, во время регрессии, у ребенка могут возникать периоды учащенного дыхания (гипервентиляция) или замедленного дыхания, включая задержку дыхания. Они также могут заглатывать воздух, что может привести к вздутию живота.
Они также могут заглатывать воздух, что может привести к вздутию живота.
Стадия 3: плато
Стадия 3 синдрома Ретта может начаться уже в возрасте 2 лет или в возрасте 10 лет. Часто это длится много лет, и многие дети остаются на этой стадии большую часть своей жизни.
На стадии 3 некоторые из симптомов стадии 2 могут исчезнуть, например, может улучшиться поведение, снизиться раздражительность и плач.
Ребенок может стать более заинтересованным в людях и их окружении, а также могут улучшиться бдительность, концентрация внимания и общение. Их ходьба также может улучшиться (или они могут научиться ходить, если раньше не могли).
Другие симптомы на этой стадии включают:
- приступы, которые могут стать более частыми
- нерегулярный характер дыхания может ухудшиться – например, поверхностное дыхание сменяется учащенным глубоким дыханием или задержкой дыхания
Также может быть сложно набрать и удержать вес.
Стадия 4: ухудшение подвижности
Стадия 4 может длиться годами и даже десятилетиями. Основными симптомами на этой стадии являются:
- развитие искривления позвоночника (сгибание позвоночника влево или вправо), известное как сколиоз
- мышечная слабость и спастичность (аномальная скованность, особенно в ногах)
- потеря способности ходить
Общение, языковые навыки и работа мозга не ухудшаются на стадии 4. Повторяющиеся движения рук могут уменьшиться, а взгляд обычно улучшается.
Судороги также обычно становятся менее серьезной проблемой в подростковом и раннем взрослом возрасте, хотя часто с ними приходится справляться на протяжении всей жизни.
Что вызывает синдром Ретта?
Почти все случаи синдрома Ретта вызваны мутацией (изменением ДНК) в гене MECP2, который находится на Х-хромосоме (одной из половых хромосом).
Ген MECP2 содержит инструкции по производству определенного белка (MeCP2), необходимого для развития мозга. Генная аномалия препятствует правильной работе нервных клеток в мозге.
Обычно синдром Ретта не передается в семейном анамнезе, что означает, что он не передается из поколения в поколение. Почти все случаи (более 99%) носят спонтанный характер, при этом мутация возникает случайным образом.
Диагностика синдрома Ретта
Синдром Ретта обычно диагностируется на основании симптомов вашего ребенка и путем исключения других, более распространенных заболеваний.
Диагноз синдрома Ретта может не ставиться в течение нескольких лет, потому что этот синдром настолько редок, что симптомы обычно не проявляются, пока ребенку не исполнится от 6 до 18 месяцев.
Генетический анализ крови можно использовать, чтобы определить, ответственна ли генетическая мутация за синдром Ретта.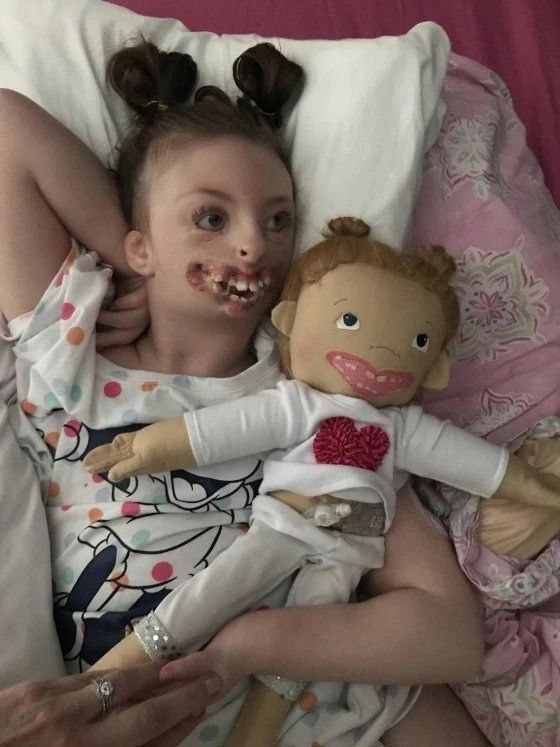 Если в гене MECP2 обнаружено изменение, это может помочь подтвердить диагноз, но его отсутствие не обязательно исключает синдром.
Если в гене MECP2 обнаружено изменение, это может помочь подтвердить диагноз, но его отсутствие не обязательно исключает синдром.
Узнайте больше о генетическом тестировании.
Лечение синдрома Ретта
Синдром Ретта неизлечим, поэтому лечение направлено на устранение симптомов.
Как родитель, ухаживающий за ребенком с синдромом, вам, вероятно, потребуется помощь и поддержка со стороны широкого круга медицинских работников.
Вашему ребенку могут быть полезны некоторые из следующих процедур и вспомогательных средств:
- речевая и языковая терапия, доски с картинками, технология взгляда и другие визуальные средства, помогающие в общении
- лекарства от проблем с дыханием и подвижностью и противоэпилептические препараты для контроля судорог
- физиотерапия, внимание к подвижности, особое внимание к сидячей позе вашего ребенка (чтобы свести к минимуму вероятность развития сколиоза) и частое изменение позы
- при сколиозе приживается, для предотвращения дальнейшего искривления позвоночника можно использовать корсет для спины, а иногда и операцию на позвоночнике (подробнее о лечении сколиоза)
- высококалорийная диета для поддержания достаточного веса с использованием зонда для кормления и других помогает при необходимости
- трудотерапия, чтобы помочь развить навыки, необходимые для одевания, кормления и других повседневных действий (они в основном используются в течение ограниченного периода времени для предотвращения членовредительства или для поощрения действий другой рукой)
- бета-блокаторы или кардиостимуляторы для контроля сердечного ритма
Лечебная верховая езда, плавание, гидротерапия и музыкальная терапия также сообщалось, что это полезно. Спросите у своей медицинской бригады, где вы можете получить доступ к этим методам лечения.
Спросите у своей медицинской бригады, где вы можете получить доступ к этим методам лечения.
Узнайте больше об уходе за ребенком-инвалидом и оборудовании для ухода, вспомогательных средствах и приспособлениях.
Outlook
Хотя некоторые люди с синдромом Ретта могут в какой-то степени сохранять способность управлять руками, способность ходить и коммуникативные навыки, большинство из них будет зависеть от круглосуточного ухода на протяжении всей жизни.
Многие люди с синдромом Ретта достигают зрелого возраста, а те, у кого болезнь протекает менее тяжело, могут дожить до старости. Однако некоторые люди умирают в довольно молодом возрасте в результате осложнений, таких как нарушения сердечного ритма, пневмония и эпилепсия.
Консультации для лиц, осуществляющих уход
Уход за ребенком с синдромом Ретта требует умственного и физического труда.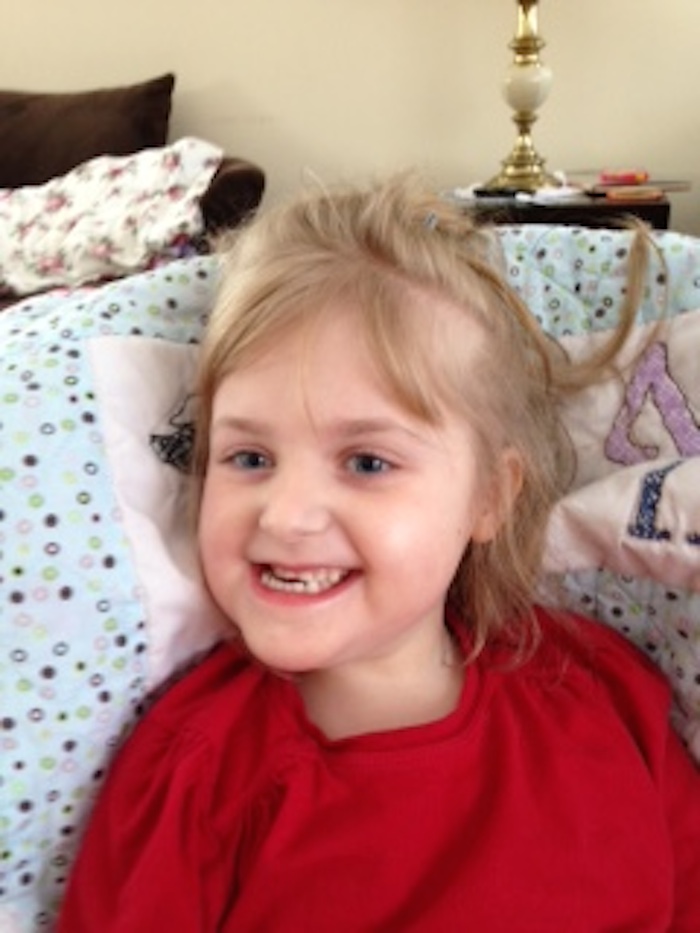 Большинству лиц, осуществляющих уход, потребуется социальная и психологическая поддержка.
Большинству лиц, осуществляющих уход, потребуется социальная и психологическая поддержка.
В вашем руководстве по уходу и поддержке содержится много информации и советов о том, как вы можете уделить время заботе о себе, в том числе:
- поддержание формы и здоровья
- перерыв от забот
- забота о своем благополучии
Возможно, вам также будет полезно обратиться в группу поддержки, например в Rett UK, за информацией и советами по уходу за ребенком с синдромом.
Национальная служба регистрации врожденных аномалий и редких заболеваний
Если у вашего ребенка синдром Ретта, ваша клиническая бригада передаст информацию о нем в Национальную службу регистрации врожденных аномалий и редких заболеваний (NCARDRS).
Это помогает ученым искать лучшие способы лечения и профилактики синдрома.
